A new technique makes it possible to track changes in gene activity across a cell’s entire genome over time in more detail than has been possible before, researchers in Seattle report. “We hope this method will improve our understanding of normal cell physiology and as well as of cell dynamics during the development of diseases or in response to drug treatments,” said lead author Junyue Cao, a postdoctoral fellow in the laboratory of Jay Shendure, professor of genome sciences at the University of Washington School of Medicine and scientific director of the Brotman Baty Institute for Precision Medicine. Shendure led the project with Cole Trapnell, associate professor of genome sciences and an investigator at the Brotman Baty Institute. Junyue Cao will start his own research group at Rockefeller University this summer. The technique, called sci-fate, was developed as part of a collaboration between the UW Medicine and Brotman Baty Institute researchers and scientists from the Allen Discovery Center for Cell Lineage Tracing in Seattle and Illumina, Inc. of San Diego, California. They describe the approach in a paper appearing April 13 in the journal Nature Biotechnology. Genes provide the instructions for proteins, which perform the work cells need done to survive. As cells grow, change and respond to challenges, some genes will become activated—or “expressed”—to produce proteins to perform specific tasks. At the same time, other genes may be inactivated or “dialed down” depending on cell’s needs. In humans, this is a continuous, dynamics involving the simultaneous up- and down-regulation of thousands of genes. Currently, scientists trying to track this complex process are taking two main approaches. One technique called “pseudotime” uses individual “snapshots” of gene expression. These are strung together in an attempt to reconstruct how expression changes over time. The other is to use a technique called time-lapse microscopy. The first technique is limited because it is necessary to infer what changes in gene expression may occur between “snapshots.” The second can track changes in only a few genes in a few cells at a time. In their new paper, the scientists show how the sci-fate technique can provide a more detailed view of gene expression changes over time, while retaining scalability to large numbers of cells. The approach combines two methods. Cells are cultured in a medium containing 4-thiouridine (4sU) an analogue of thymidine, one of the four bases that make up RNA. A form of RNA, called messenger RNA, carries the instructions encoded in the genes to the cellular machinery that synthesizes proteins. As a result, when genes in the cells cultured in this medium are activated, 4sU is incorporated into new mRNA. The 4sU thus serves as a marker that identifies which mRNA in the cells are new, therefore indicating which genes are currently being expressed and by how much. This process of converting a gene’s instructions from DNA to RNA is called transcription. The total of mRNA transcripts in a cell is called its transcriptome The second key to the new technique is called single cell combinatorial indexing, the “sci” in sci-fate. In this process, individual cells and mRNA transcripts within the cells are molecularly “barcoded” to allow the scientists to identify which individual cell the mRNA transcripts originate. To demonstrate the approach, the scientists took more than 6,000 single cells derived from a lung cancer. Such cells are often used in research because the cells are essentially immortal and so can be cultured year after year for study. The cells were cultured in the 4sU medium. To show how the technique could track alterations in cell function, the cells were treated with a powerful drug that mimics the effect of the hormone cortisol, called dexamethasone. The scientists chose this drug because cortisol activates proteins that bind to thousands of sites on the genome. This activation rapidly triggers significant changes in gene expression. The researchers then analyzed the transcriptome of the cells without exposure to dexamethasone and after 2, 4, 6, and 10 hours after exposure. By using this approach and by applying computational analyses, they were able to compare transcriptomes of the different cells. These comparisons allowed them to construct a detailed sequence of the changes in gene expression — which genes were being up-regulated and which were being down-regulated simultaneously across the cell’s genomes — as the cells went through their normal cell cycle and responded to the dexamethasone treatment. “We anticipate this method will allow scientists to track, measure and characterize changes in gene expression and in a wide variety of cell systems,” said Shendure. Please see the Nation Biotechnology paper, “Characterizing of the temporal dynamics of gene expression in single cells with sci-fate.” This work was supported by the Paul G. Allen Frontiers Foundation, National Institutes of Health (DP1HG007811, R01HG006283), W. M. Keck Foundation, Dale. F. Frey Award for Breakthrough Scientists, Alfred P. Sloan Foundation Research Fellowship, Howard Hughes Medical Institute, and Brotman Baty Institute for Precision Medicine.
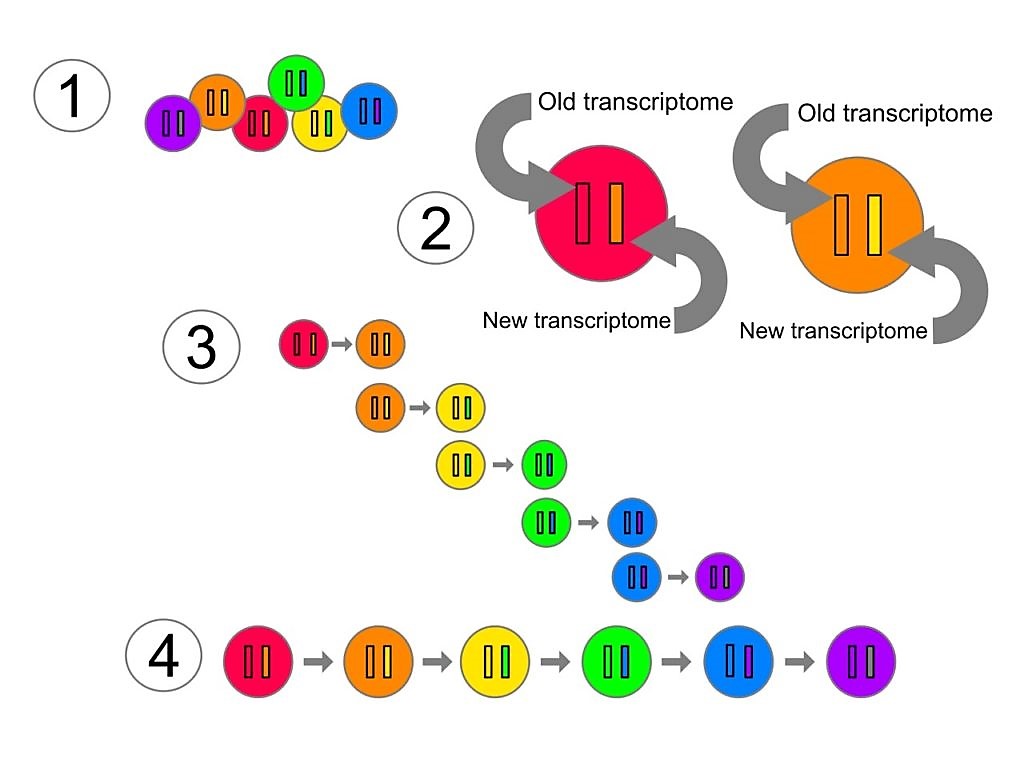
Explanation of sci-fate illustration
- Cells are cultured in a marker that is taken up into new mRNA transcripts when they are synthesized.
- Since the old transcripts do not contain the marker, it is possible to distinguish new from the older transcriptomes.
- By comparing the transcriptomes in the cells, it is possible to determine which cell transcriptomes represents an earlier and which a later state.
- Using such comparisons, it is then possible to reconstruct a single cell’s trajectory through its cell cycle and response to dexamethasone.